Molecular dynamics simulation of receding and advanced contact angle dynamics
Sergii Burian and Volodymyr Kovalchuk, Institute of biocolloid chemistry named after F.D. Ovcharenko of National Academy of Sciences of Ukraine. These results are a part of studies performed under the European Commission project “EHAWEDRY” within the scope of FP Horizon 2020 (Grant Agreement No. 964524)
The principal possibility of energy harvesting by mechanical actuation of the contact area between electrolyte solutions and hydrophobic electrode surfaces has been rather convincingly demonstrated. An increase of the generated power density should be possible by employment of porous electrodes with very fine pores. As the mechanical actuation is problematic with such electrodes, dewetting via solvent evaporation could be a solution. In this case moderately hydrophobic surfaces, which can be wetted spontaneously, are more suitable. However, a thin wetting film remaining after the receding meniscus (or formed by adsorbed solvent molecules) may prevent obtaining perfectly dry electrode surfaces. Formation of such wetting films is possible in the case of positive disjoining pressure stabilizing the film. In spite of intensive experimental and theoretical studies of such films, there is no complete understanding of the origins of their formation, in particular, on the surface of conductive materials with different hydrophobicity.
To address the problem of receding and advancing contact angle dynamics and possible wetting film formation we conducted molecular dynamic numerical simulations. To simplify the task, as a first step, we conducted simulations for pure water drop displacement over silicon surface. Silicon is widely employed in various simulations, what allows using of well-known interaction parameters for silicon atoms and water molecules. Silicon can become conductive if doped with electron donor or acceptor impurities. This was, however, not employed in the present simulations.
Details of the simulation of a water drop on a silicon substrate
All numerical calculations by the classical MD method were made using the open code LAMMPS (Largescale Atomic/Molecular Massively Parallel Simulator) and implemented mainly on the computer cluster of the LEMTA laboratory (a joint laboratory of French National Centre for Scientific Research and the University of Lorraine) and partly on the computer station of the Department of Macrokinetics of Natural Disperse Systems of F. D. Ovcharenko Institute of Biocolloidal Chemistry of the National Academy of Sciences of Ukraine.
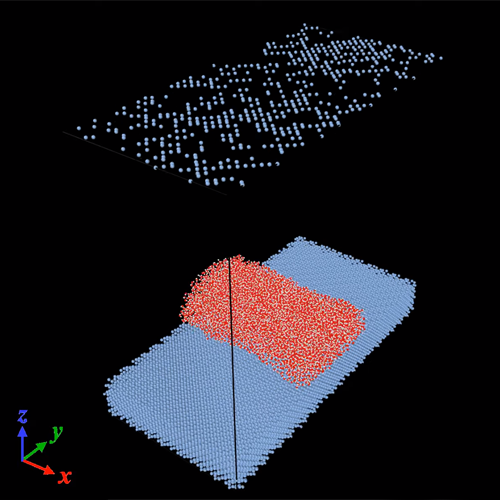
The results of the MD modeling of a cylindrical water droplet placed on an atomically uniform silicon substrate are presented in this Section. The stability of the ‘cylindrical sessile nanodroplet’ was ensured by the establishment of periodic boundary conditions (in the simulation box along the x-axis), that is, by its infinite periodic extension along its axis of translational symmetry. The water droplet interacts only with the silicon surface (001) without the influence of the external field at the first stage of the simulation. All silicon atoms were free to move, so only thermally vibrating surfaces were considered, since Barisik and Beskok proved the fixed silicon surface is not a correct model representation.
We considered three types of interparticle interactions in computer simulations, namely between water molecules, between silicon atoms, and between silicon atoms and oxygen. We neglected the interaction between hydrogen and silicon atoms. The first type of interactions (between water molecules) is modeled using the SPC/E water model (Simple Point Charge/Extended). Parameters for describing this model are presented in several works. The interaction of silicon atoms is described by the Stilling-Weber potential. The third type of interactions (between silicon and oxygen) is described by the Lennard-Jones interatomic potential, with the following parameter values: Si-O ε = 15.61 meV and σ Si-O = 2.6305 Å. A harmonic force was also applied to the silicon atoms of the lower four atomic layers in order to bind them to their initial positions and thereby to avoid the moving of the silicon substrate in the z-axis direction during the simulation. Velocity Verlet algorithm was used to numerically integrate of Newton’s equations with a step of 1 fs. The sizes of the simulation box, the number of atoms of the silicon substrate and the cylindrical droplet are presented:
Simulation box: 108.6 Å×271.5 Å×200.0 Å
Silicon substrate: 108.6 Å×271.5 Å×43.44 Å
Initial configuration of a water droplet: 108.6 Å×43.4 Å×43.4 Å
Number of Silicon atoms: 4×20×50×8 = 32 000
Number of Water atoms: 3×35×14×14 = 20 580
All atoms of the system were given initial velocities after creating the initial configuration, which corresponded to an average temperature of 1 K. Then this system was ‘heated’ from 1 K to 300 K for 0.3 ns, and for 1.7 ns, its thermalization continued at a temperature of 300 K. The Nosé–Hoover thermostat in the canonical ensemble was used for this. An equilibrium and stable drop in the form of a cylindrical segment was obtained as a result of this thermosetting (see Video 1 and Video 2).
Modeling a droplet under the influence of an external field
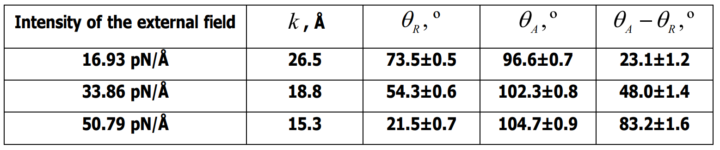
At the second stage, a series of three simulations of the previously obtained equilibrium system was carried out, but under the influence of an external uniform force field of varying intensity. The force field was oriented parallel to the surface of the silicon substrate along the y-axis. Steady-state receding and advancing contact angles at three different values of the external field intensity (16.93 pN/Å, 33.86 pN/Å and 50.79 pN/Å) were obtained (see below).
As a result of simulations, it was found that the droplet under the influence of an external field first moves with acceleration for some time, not exceeding 1 ns, after which its motion is practically uniform. The fact, that the droplet acquires uniform movement over time, makes possible to subtract the y-component of the velocity of the center of mass of the vapor-liquid system (the droplet together with its saturated vapor) from the corresponding component of the velocity of the center of mass of each atom of the vapor-liquid system in numerical calculations. Thus, the reference frame can be chosen in MD calculations, relative to which the droplet will be stationary, and only the thermal motions of water molecules remain. The droplet displacement under the action of an external field is illustrated in Video 3.
The possibility of fixing the center of mass of the vapor-liquid system by the specified procedure reduces the problem of the shape establishing of the droplet profile based on the density distribution map. Therefore, the three-dimensional space of the simulation box was divided into equal straight parallelepipeds with dimensions of 108.6 Å×2 Å×2 Å and the average values of the density in each such cell were calculated. Droplet shapes were determined using these averaged values.
The values of the advancing and receding angles for three different values of the external field intensity, which are shown below, were obtained by the method of numerical adjustment of the parameters of the developed nonlinear model:
Watch the Simulated Videos:



The results of MD simulations were visualized by using the OVITO software (A. Stukowski, Visualization and analysis of atomistic simulation data with OVITO – the Open Visualization Tool, Modelling Simul. Mater. Sci. Eng. 18 (2010), 015012).